
Experimental Design and Library Prep
The first step of an RNA-Seq project is the definition of the experimental design, which itself depends on a scientific question. For instance, an oncologist gaining insight on the effect of a certain chemotherapeutic agent may want to determine which genes are up or down regulated in the liver of cancer patients treated with the compound.
Once the scientific question has been defined, next step is setting up the RNA experiment. An important part of this process is determining the number of biological and technical replicates. While technical replication is often unnecessary, mainly due to the robustness of current protocols, biological replication deserves thoughtful consideration. Whether the experiment is performed on cell-lines, in-vivo model organisms or humans will influence the minimum number of replicates. Every experiment is unique and we can assist on defining the number of biological replicates.
Next step is RNA extraction and sample delivery. Extraction is performed at the researcher’s lab and brought to our premises personally or by courier. In order to identify the samples, a GC code label has to be attached to each of them. GC codes are requested in our website when the project is registered. Each sample must have assigned a unique GC code.
 After all samples are received, they are prepared for sequencing. For differential expression analysis, Illumina’s TruSeq Stranded mRNA and QuantSeq 3’ mRNA-Seq library prep Kits are available. If any of the samples presents a quality issue before or during library preparation, the user is contacted by our technicians.
After all samples are received, they are prepared for sequencing. For differential expression analysis, Illumina’s TruSeq Stranded mRNA and QuantSeq 3’ mRNA-Seq library prep Kits are available. If any of the samples presents a quality issue before or during library preparation, the user is contacted by our technicians.
Sequencing
As soon as the library preparation is complete, the samples are scheduled for sequencing. Read length, single or paired-end sequencing and minimum number of reads per sample are important parameters:
- Read length refers to the number of bases each read has. For differential expression analysis typical number is 50, but others lengths are possible as well. In general, the longer the read the lower the chance it will map to more than one location in the reference genome. The trade-off is that producing longer reads requires more sequencing cycles, which increase costs.
- Single or paired-end refer to whether the fragments are sequenced from one or both ends. Paired-end sequencing improves mapping, but it increases costs. In typical differential expression experiments, single-end sequencing is sufficient.
- A minimum number of reads per sample is required in order to have enough evidence for making a call between conditions.
Read length, pairing strategy and number of reads are all discussed and agreed with the user during the experimental design meeting.
In our facility, sequencing of RNA-Seq projects is typically performed on Illumina’s HiSeq4000, HiSeq2500 or NextSeq systems.
After a period of typically six weeks from the moment the samples are delivered, sequencing is complete and the scientist assigned to the project takes contact with the user to discuss the sequencing results and the bioinformatics analysis.
Bioinformatics Analysis
The bioinformatics analysis to identify differentially expressed genes consists of essentially three steps: mapping, feature counting and statistical inference.
- Raw reads stored as FASTQ files are quality filtered and then mapped to the reference genome/transcriptome with the aid of a splice-aware aligner. The output is a BAM file.
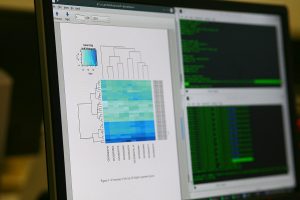
- Read counts is correlated to the abundance of the target transcripts. The output of this step is a table which reports, for each sample, the number of reads that have been mapped to a gene/feature.
- Statistical inference to detect differentially expressed genes.
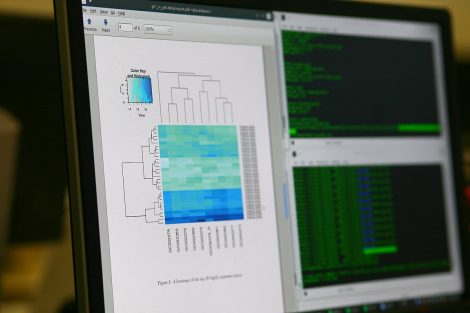
When the analysis is complete, the user is invited to discuss the results. The data transfer is performed either via hard drive or the secure cloud.
Resources
RNA-Seq and experimental design
RNA-Seq: a revolutionary tool for transcriptomics
RNA-Seq Data Analysis – a Practical Approach
Library preparation
QuantSeq 3′ mRNA-Seq Library Prep Kit FWD for Illumina
Bioinformatics
Systematic evaluation of spliced alignment programs for RNA-seq data
A survey of best practices for RNA-seq data analysis
Contact
If you would like to discuss your RNA-Seq project with us, please contact us [email protected].